






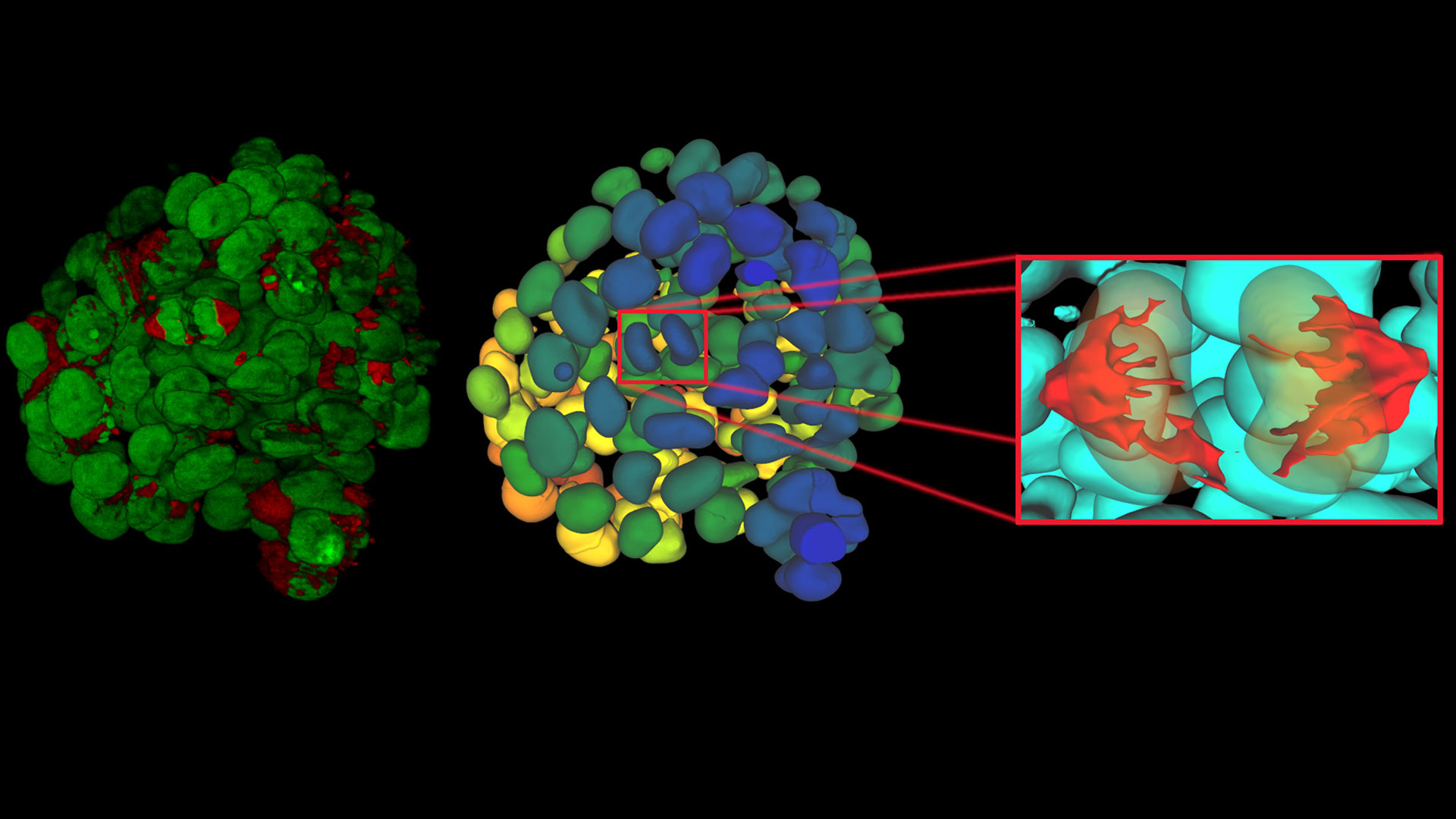





, insulin SGs (orange), microtubules (red), nucleus (yellow), and plasma membrane (transparent).")





, tubulin with Cy5 (red), and nuclei with DAPI (blue). Image courtesy of Dr. Günter Giese, Max Planck Institute for Medical Research, Heidelberg, Germany.")


, Tropomyosin (cardiomyocytes, red) and GFP (primordial cardiac layer, green).")


Leica Microsystems launches the Viventis SCAPE light sheet microscope
Our Latest Articles
High-Pressure Freezing Protocols for Ultrastructural 3D EM
High pressure freezing (HPF) can help preserve hydrated cells and tissues close to their biological state at the moment of immobilization, supporting more reliable ultrastructural interpretation than…
Ultramicrotome UC Enuity in Practice: Stable 15 nm Sections at ZFE
After using the UCT and UC6 ultramicrotomes, Claudia Mayrhofer calls UC Enuity a leap in stability—so robust that vibrations and temperature shifts don’t spoil sections, even with multiple users. Auto…
Ensuring Glass Quality with the Polarization Microscopy Advantage
Glass is one of the oldest materials known. Today, it is used for many applications, e.g., optical instruments, windows, doors, solar panels, containers for food, beverages, and medicine, so strict…
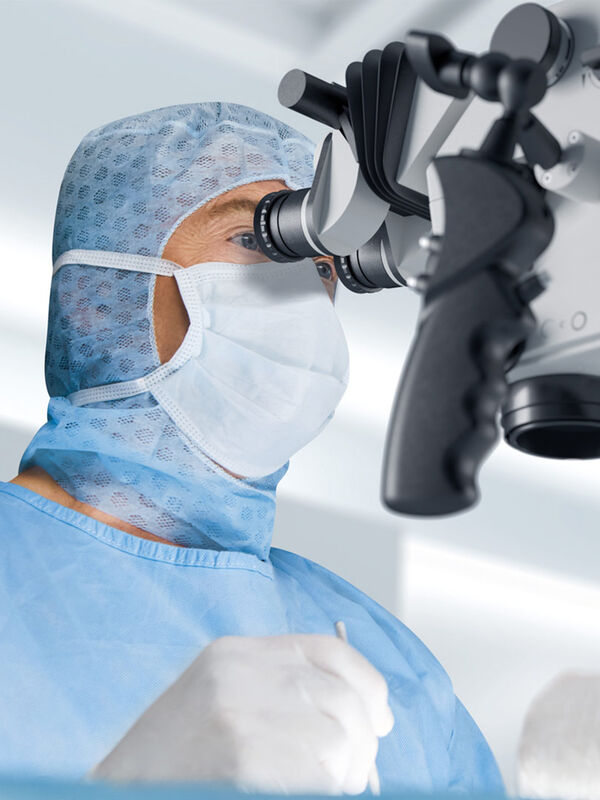
Expert Techniques for Superior Visualization in Cataract Surgery
Join renowned ophthalmic surgeons, Dr. Hussein Almuhtaseb and Mr. Simon Madge, as they share their clinical expertise and real-world surgical strategies during the 2025 Online Cataract Surgery…
Eliminating Electrostatic Interference in Laser Microdissection
Electrostatic charge in laser microdissection (LMD) causes two critical failures: samples stick to charged surfaces and are lost, or samples fly into adjacent wells and cause cross-contamination. We…
History, Developments and Trends of Microscopy in Cancer Research
Cancer is a global disease, with 18 million new cases diagnosed and 10 million cancer-related deaths worldwide in 2020. This burden is set to increase, with a projected increase in cases of ~55% by…
Overview of Fluorescent Dyes in terms of Applications and Properties
An introduction to commonly used fluorescent dyes and an overview of their characteristics are given in this article. Fluorescence microscopy is used for the study of specific cellular components with…
Researchers Insights: Microscopy in Cancer Research
Discover how imaging techniques are driving cancer research forward. In this issue, we present comprehensive multimodal studies using microscopy, as well as new directions in intraoperative cancer…
Predictive Service Prevents Downtime in Ghent
At the VIB BioImaging Core in Ghent, Belgium, researchers depend on Leica’s Stellaris 8 confocal microscope to explore the frontiers of biomedical science. When Leica’s RemoteCare system detected a…
A Guide to Fluorescence Microscopy
Fluorescence microscopy uses the ability of fluorophores, dyes, or fluorescent proteins to emit light of a specific wavelength after being excited with light of a shorter wavelength. Biomolecules can…
ATTO-TEC Consumables
ATTO-TEC dyes have become a benchmark for fluorescence microscopy imaging, offering a highly differentiated panel. Their brightness and photostability make them the reagents of choice for demanding applications.

February 2026, Wetzlar, Germany - Leica Microsystems, a Danaher company and a leading provider of microscopy and scientific solutions, has appointed…

Leica Microsystems CEO, Dr. Annette Rinck, accompanied German Federal Chancellor Friedrich Merz as part of the German economic delegation during the…

Leica Microsystems appoints Diamond Dent Trading Co as Authorized Partner for Dental Surgical Microscopy Solutions in Saudi Arabia

Convenient access to tailored microscopy solutions via AI-powered search
